Replacing Lab Experiments with Computation
Fast, accurate AI physics simulations for pharma. Compute solubility, lipophilicity, crystal structure stability and more with accuracy matching experimental error.
Get in touchTeam
-

Laurence Midgley
Cofounder, CEO


-

Miguel Hernández-Lobato
Cofounder, Head of AI
-

Gábor Csányi
Cofounder, Head of Science
-

Bálint Máté
Member of Technical Staff
-

Graeme Day
Advisor
Our Science
arXiv, 2026 DFT Accuracy on Crystal Structure Prediction with Machine Learning Interatomic Potentials
In collaboration with 
DFT Accuracy on Crystal Structure Prediction with Machine Learning Interatomic Potentials
We present an evaluation of CSP-MACE-Å, a machine learning interatomic potential intended to replace DFT in crystal structure prediction (CSP). By running multiple orders of magnitude faster than DFT, CSP-MACE-Å enables energy and free energy evaluation of far more candidate structures, providing greater confidence when derisking solid forms.
 Read paper →
Read paper →
We introduce an efficient alchemical free energy protocol that enables calculations of rigorous free energy differences in condensed phase systems modeled entirely by MLPs. Using a pretrained, transferable, alchemically equipped MLP model, we demonstrate subchemical accuracy for the solvation free energies of a wide range of organic molecules.
 Read paper →
Read paper →
We introduce FAB, a new method for training generative models to sample accurately from Boltzmann distributions of molecular systems using only the energy function, without requiring samples from the target distribution. We are the first to learn the Boltzmann distribution of the alanine dipeptide molecule using only the unnormalized target density, without access to samples generated via Molecular Dynamics (MD) simulations.
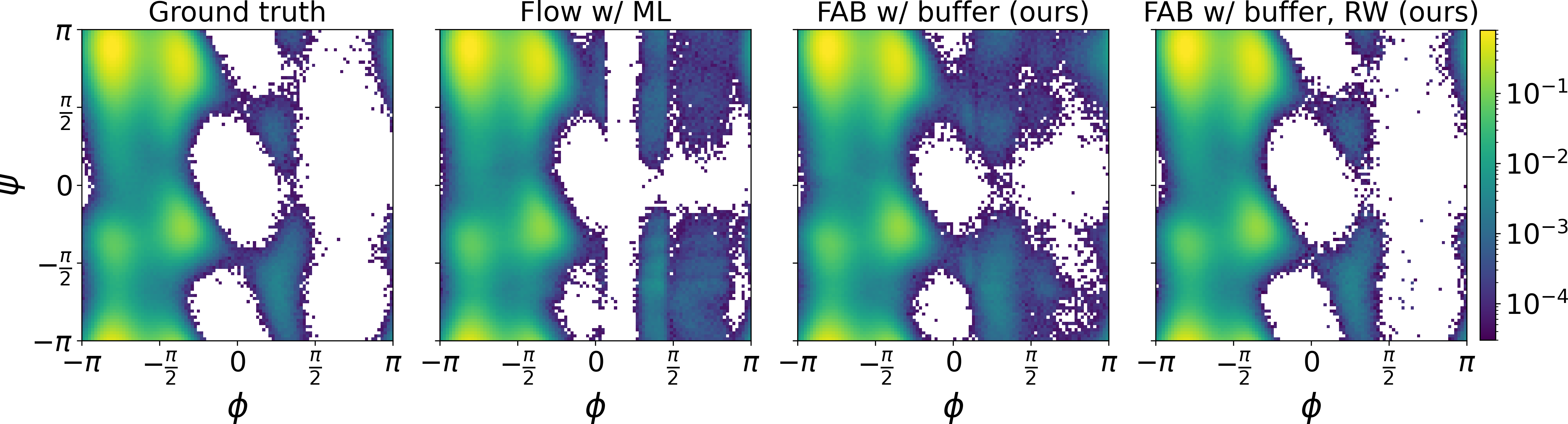 Read paper →
Read paper →
We present a coupling normalizing flow architecture that preserves SE(3) and permutation equivariance while providing both fast sampling and density evaluation. We are the first to learn the full Boltzmann distribution of alanine dipeptide by only modeling the Cartesian positions of its atoms.
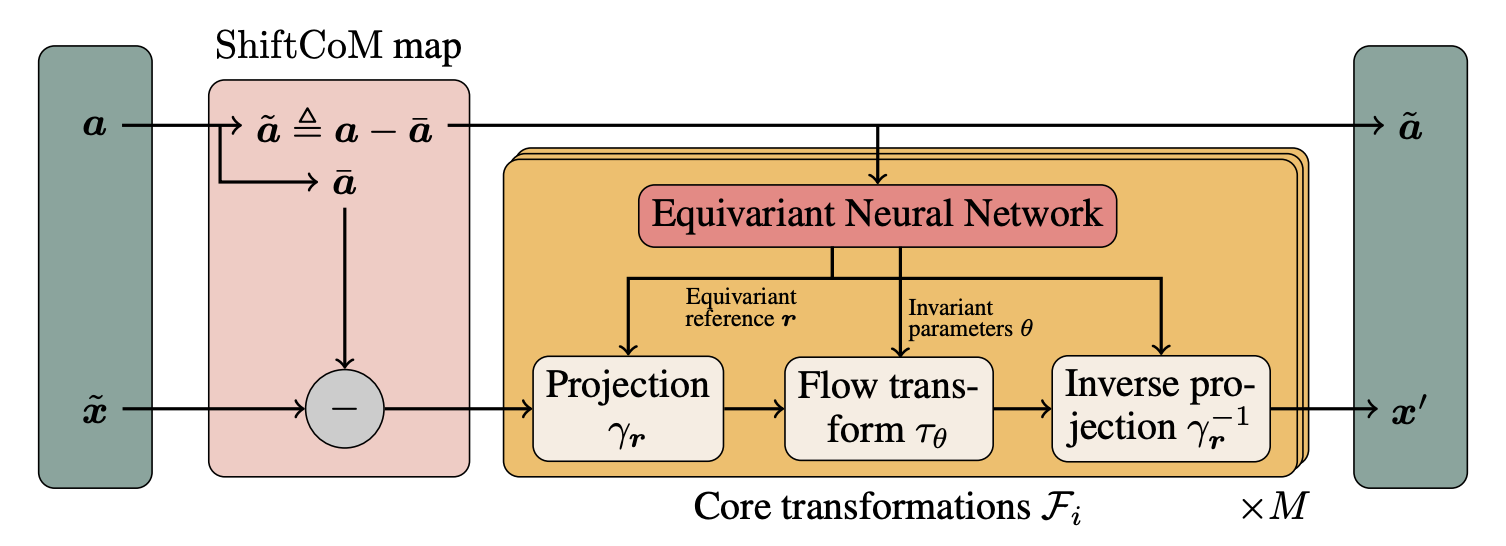 Read paper →
Read paper →
Reach out to us
Interested in seeing what Ångström AI can do for your drug discovery pipeline? We'd love to show you a demo.
info@angstrom-ai.com