Replacing Lab Experiments with Computation
Fast, accurate AI physics simulations for pharma. Compute solubility, lipophilicity, crystal structure stability and more with accuracy matching experimental error.
Get in touchTeam
-

Javier Antorán
Cofounder, CEO


-

Laurence Midgley
Cofounder, CTO
-

Harry Moore
Lead Computational Chemist
-

Miguel Hernández-Lobato
Cofounder, Chief AI Officer
-

Gábor Csányi
Cofounder, Chief Scientific Officer
Our Science
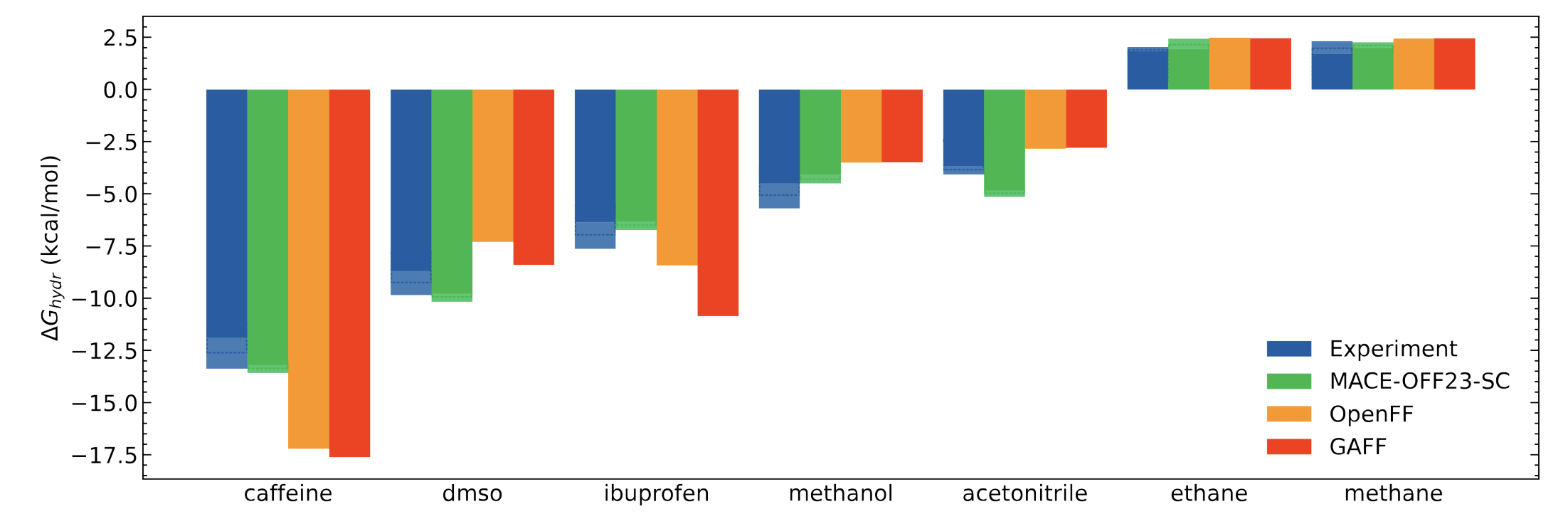
An alchemical free energy method that achieves sub-chemical accuracy for the hydration free energies of organic molecules, combining machine learning force fields with rigorous statistical mechanics.
 Read paper →
Read paper →
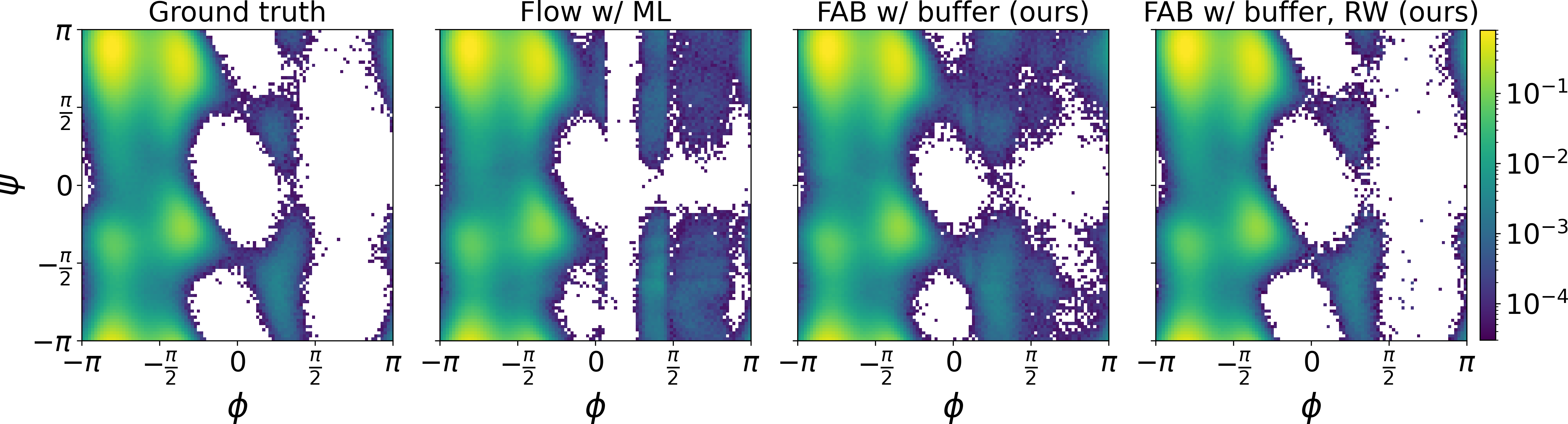
A method for training generative models on Boltzmann distributions using only molecular energy functions, without requiring samples from the target distribution. Enables accurate sampling of complex molecular systems.
 Read paper →
Read paper →
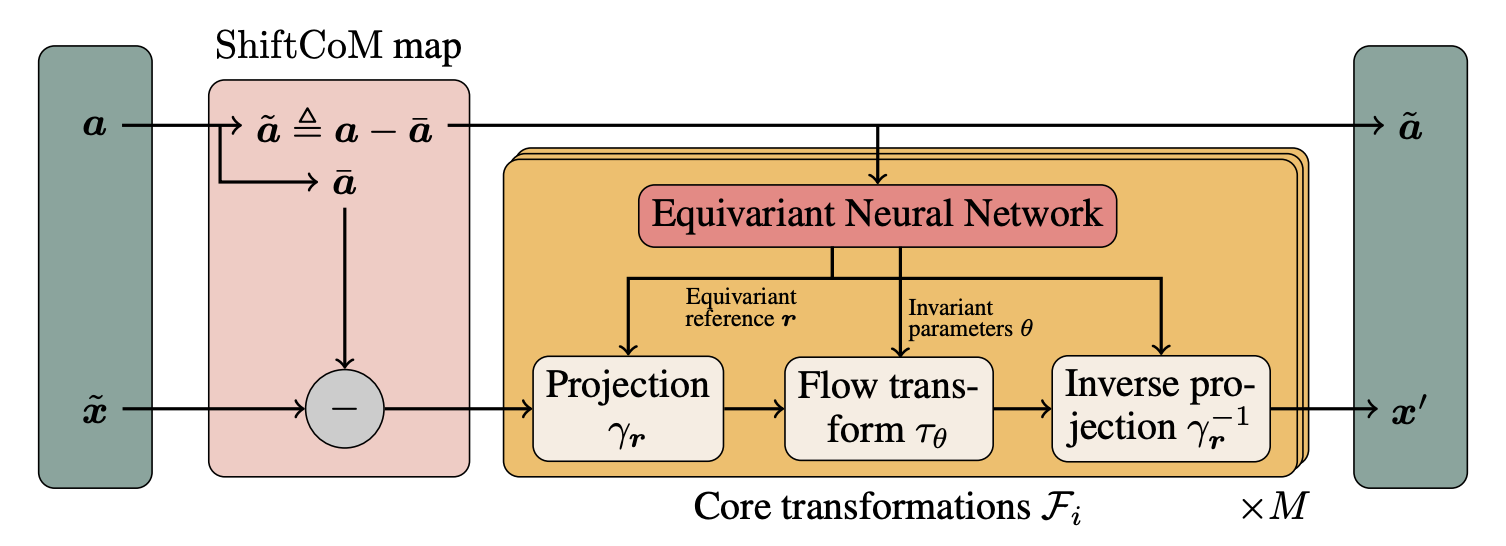
An equivariant generative model for molecular conformations that respects the physical symmetries of 3D space. Achieves sampling more than an order of magnitude faster than traditional molecular dynamics.
 Read paper →
Read paper →
Reach out to us
Interested in seeing what Ångström AI can do for your drug discovery pipeline? We'd love to show you a demo.
info@angstrom-ai.com